

Research in Biochemistry
Studying the Potential Applications of Dipeptide Nanomaterials
By Lisa Perreault
I am a senior biochemistry major at UMass Dartmouth, pursuing the 4+1 BS/MS program degree path. In addition to being a full time chemistry student, I am a chemistry teaching assistant and an undergraduate student researcher. Since the spring semester of 2015, I have been involved with the Mayes Research Group, which focuses on computational and theoretical chemistry. During my time in the group, I have been working on a dipeptide nanotube modeling project, which is centered on the self-assembly of this innovative nanomaterial. This research was partially funded through a grant from the OUR. All of our calculations run on the Massachusetts Green High Performance Computing Center (GHPCC), a state wide computing cluster with high computing capabilities. Using the GHPCC allows my calculations to be carried out quickly and efficiently, while teaching me a unique set of computing skills that not many undergraduates get to learn. After graduation, I hope to take what I have learned at UMass Dartmouth as a student researcher and apply it to a career in pharmaceutical drug development.
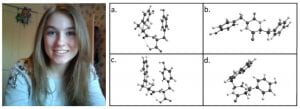
Left: Portrait of Perreault; right:The molecular structures of (a.) linear YY, (b.) cyclic YY, (c.) linear WY, and (d.) cyclic WY
Dipeptide nanomaterials are a relatively new and unique biomaterial with many potential applications. Their organic nature, rigidity and flexibility make them safe, yet strong, lending them to biological applications, such as biosensing, tissue engineering, and biological scaffolds. Their semiconductor properties make them potential alternatives for electrical materials, such as solar cells. During the past several years, these dipeptide nanomaterials have risen in scientific interest and their properties have been investigated on a both a macro and micro scale. However, much is still unknown about the self-assembly of these dipeptide nanostructures. The aim of my research is to investigate the self-assembly of aromatic dipeptide nanotubes, using a variety of quantum computational methods. Four dipeptides are considered in my research: linear dityrosine (YY), cyclic YY, linear tryptophan-tyrosine (WY), and cyclic WY.
The basic theorized mechanism of nanotube self-assembly is that monomers form small aggregates, which then form rings, which stack to form tubes. So far, a bottom-up approach has been used to model the initial steps of nanotube self-assembly in order to study the fundamentals of the process. Progress so far can be broken down into three basic stages: a study of each of the four dipeptides, a study of their dimers, and a study of hexamer rings made from these dipeptides.
In the first stage of the study, the exact structure and energetics of linear YY, cyclic YY, linear WY, and cyclic WY were determined. Spartan software was used to determine all of the geometrically and energetically favorable conformations of each dipeptide. From there, the fifteen lowest energy conformers were analyzed further using GAMESS (General Atomic Molecular and Electronic Structure) to determine more accurately the lowest-energy conformer of each dipeptide, representing their most stable form. Geometric, molecular orbital, and IR spectra calculations were also performed to analyze the molecular trends present in low-energy conformers. The most important similarity between the most stable conformers of the four dipeptides is their stabilizing interactions. Each dipeptide has a relatively high dipole moment, implying that there are important polar interactions involved in their stabilization. Additionally, each dipeptide is characterized by highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) consisting of mainly π and π* orbitals, respectively, suggesting that π-π* stacking interactions are also important. Finally, the addition of acetone solvation lowered the energy of each conformer, suggesting the solution stabilized the dipeptides and stimulates self-assembly experiments.
 Left: Examples of the (a.) side by side, (b.) stacking, (and c.) T orientations studied, using the cyclic YY dipeptide; right: An example of the hexamer ring, using the linear YY dipeptide
Left: Examples of the (a.) side by side, (b.) stacking, (and c.) T orientations studied, using the cyclic YY dipeptide; right: An example of the hexamer ring, using the linear YY dipeptide
In the second stage of the study, dimers of each of the four dipeptides were studied. The lowest energy conformers from stage one were dimerize in three orientations: a linear “side by side” interaction, a parallel “stacking” interaction, and a perpendicular “T” interaction. GAMESS was used to optimize and analyze the dimers. Again, the geometries, energetic properties, molecular orbitals, and IR spectra of each system were investigated. Analysis of these properties showed that the stacking interactions have the overall lowest energy, the greatest binding energy, the most hydrogen bonding between dipeptides, and the tightest packing of the dipeptides. This suggests that dipeptides have a tendency to stack above one another in the early steps of their self-assembly.
To investigate the interaction between the dimers even further, an energy decomposition calculation was carried out in GAMESS on each dimer. This calculation computes the types and amounts of interactive forces present between two molecules. It showed that the dominant interacting force in each of the dimers was electrostatic energy (accounting for ~50% of the total interaction energy) and polarization energy (accounting for ~30% of the total interaction energy). This implies that the dipole-dipole interaction between peptide bonds and the non-covalent interactions of the peptide termini play an important role in the interactions between multiple dipeptides.
In the third stage of the study, hexamer rings of each of the dipeptides were studied. The lowest-energy conformers from stage one were arranged into six-membered rings and optimized using GAMESS. Again, the geometric, energetic, orbital, and IR properties were analyzed. The binding energies were calculated to be moderately large, suggesting that the dipeptides have high affinity for each other in this ring arrangement. Linear YY and linear WY have binding energies nearly twice as large as those of their cyclic counterparts, suggesting that they will self-assemble more readily than cyclic YY and cyclic WY. The inner and outer diameters of each ring were calculated and compared against experimental data for the highly studied diphenylalanine nanotube, revealing that these four nanotubes will be slightly larger, due to large side chains and higher polarity.
§
I hope to continue working on this project for the rest of the academic year at UMass Dartmouth. The project can take several directions from here. This includes combining the rings into stacks to model complete nanotubes and performing molecular mechanics calculations on the large system to determine if any new molecular interactions arise in the nanotube system. It will also shows the interactions that occur between a field of nanotubes. Another possible trajectory is to model and study the mechanical properties of the nanotubes to reveal their strength and flexibility, which would be important for applications. The path that I will choose to study first is the interaction of these dipeptides with surface materials. This also has implications for the application of dipeptide nanotubes; it shows if the nanotubes will be compatible with the surfaces. Above all, it demonstrate the ways in which the nanotubes interact with these surfaces.